Produced in Partnership with RNAConnectReviewed by Maria OsipovaOct 10 2025
The UltraMarathonRT® cDNA Synthesis and Amplification Kit delivers a comprehensive approach for copying polyA+ RNA into cDNA amenable to several later uses and analyses. The resulting PCR-amplified full-length cDNA is appropriate for use in transcriptomics library prep and cDNA library cloning as well as Rapid Amplification of cDNA ends (RACE).
The enzymatic engine powering the UltraMarathonRT (uMRT) kit is an ultra-processive Group II-intron-derived reverse transcriptase (RT) built to copy RNAs of different lengths, such as long and structured RNAs, from end to end in a single pass.
This enzyme offers several advantages over retroviral (MMLV & AMV alike) RTs, for example, improved processivity, intrinsic helicase activity to naturally unravel secondary and tertiary RNA structures, and high polymerase activity at reduced temperatures (30 °C), key for maintaining RNA sample integrity.
As uMRT reliably copies RNA from 3’ to 5’ ends irrespective of length, structure, or complexity, RNA-seq libraries produced via this synthesis are:
- A 10-30 % increase in the number of genes identified
- Gene quantification that is more reliable, especially for longer transcripts
- Reduced bias due to superior gene body coverage
- Greater diversity of isoforms
- New biology from transcripts previously not detectable from retroviral RTs
Both the components and the workflow of the uMRT are built to produce first strand cDNA products tagged with a known oligonucleotide sequence on both termini, making it possible for them to be amplified by PCR. This happens in a single tube via two sequential reactions, streamlining the process with UltraMarathonRT’s powerful extension and template switching activities (Fig. 1).1
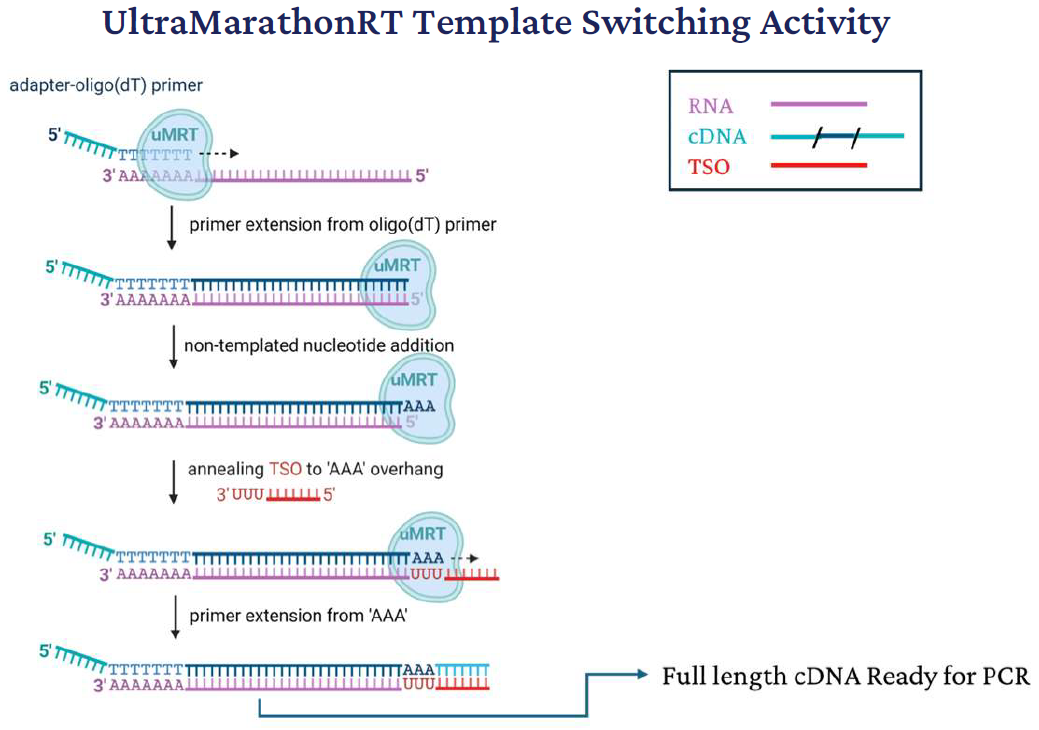
Figure 1. Mechanism of template switching by UltraMarathonRT (uMRT). Image Credit: Guo LT. RNA. 2024 Oct 16;30(11):1495-1512
Figure 1 shows the mechanisms of template switching. uMRT initiates reverse transcription from an adapteroligo( dT) primer annealed to the poly(A) tail of RNA. Following cDNA synthesis, uMRT adds several non-templated adenines at the 3′ end of the nascent cDNA. This overhang facilitates annealing of a template-switching oligonucleotide (TSO) containing a complementary 3′ rGrGrG sequence.
uMRT then extends the cDNA using the TSO as a template, incorporating the adapter sequence into the cDNA product. This process enables the generation of full-length cDNA molecules with defined 5′ and 3′ adapters for downstream applications.
For ease of use, the workflow is “addition-only,” where novel parts are added to the reactions so that no further cleanup steps are needed until after the last PCR amplification (Fig. 2).

Figure 2. UltraMarathonRT cDNA Synthesis and Amplification Kit workflow. The streamlined workflow uses an addition-only format where uMRT performs reverse transcription and template switching in under one hour, followed by PCR amplification and bead purification. The process generates high-quality full-length cDNA libraries suitable for downstream sequencing. Image Credit: RNA Connect
In the initial reaction, an adapter-tagged oligo-dT primer is annealed to polyA tails and made longer by UltraMarathonRT to copy the polyA+ RNAs into full-length cDNA. After this, the parts for template switching are administered to the reaction to enable proper tagging at the cDNA’s 3’ end. To switch templates, non-templated nucleotides are added to the 3’ end of the cDNA before annealing the provided template switching oligo (TSO) and copying of the TSO.2
Both UltraMarathonRT reactions, extension, and template switching are optimized to ensure the workflow’s sequential nature provides the best results. The cDNA produced is then PCR amplified with the included parts and cleaned using magnetic beads to produce double-stranded full-length cDNA appropriate ready for short-read sequencing library prep. A representative Bioanalyzer trace of the uMRT cDNA size distribution against two retroviral kits can be seen in Fig. 3.
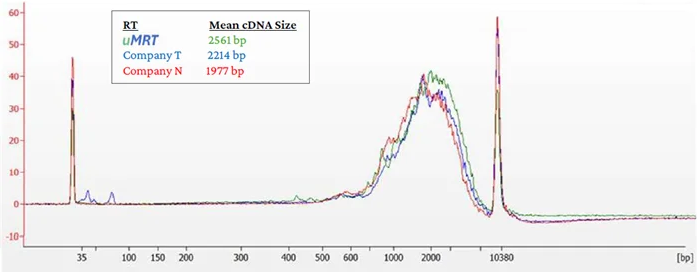
Figure 3. cDNA QC: Bioanalyzer traces depict the double-stranded cDNA size distribution after PCR amplification and magnetic bead purification. The mean size from each kit is shown with colors matching the trace. Image Credit: RNA Connect
The PCR guidelines deliver well-amplified full-length cDNA for input into various short-read library prep approaches that start with enzymatic fragmentation or tagmentation with transposase. The UltraMarathonRT cDNA Synthesis and Amplification Kit was benchmarked against two top commercially-available cDNA synthesis and amplification kits using Universal Human Reference RNA (UHRR), a pooled group of 10 human cancer cell lines which are typically used for benchmarking samples and short-read sequencing workflow.
All three of the kits use similar steps in molecular biology that produce cDNA through template switching after PCR, but the comparator kits contain a retroviral RT. Spike-in RNAs, such as ERCC and long SIRV transcripts (SIRV-Set 4, Lexogen) were included as a standard. These polyA+ RNAs cover a broad spectrum of RNA lengths and masses.
Following cDNA synthesis and template switching, each of the samples were amplified with PCR. The purified cDNA was then transformed into a short read library with the Illumina Nextera XT DNA Library Preparation Kit. Illumina pair-ended short-read sequencing on the Novaseq (2x150 bp) was conducted to a depth of 18M-22M read pairs for each sample. The sequencing reads were then down sampled to 18 M read pairs for a gene detection analysis.
Transcriptomics: Contrasting uMRT with Retroviral RTs
The RNA-Seq data sets were matched to the human genome (GRCh38.p14) and to reference transcriptome databases (Gencode annotation v47 for gene and intron detection and NCBI curated annotation for genebody coverage analysis).
The outcomes mapped data from the UltraMarathonRT cDNA Synthesis and Amplification Kit displayed a higher degree of gene detection, gene body coverage, and better detection of introns. Moreover, analysis of the spike-in controls demonstrated that UltraMarathonRT achieves superior RNA quantification than the two retroviral kits.
Detecting Genes
Better gene detection was seen across all kinds of RNA species with long noncoding RNAs displaying the most significant increase relative to the retroviral RT data sets; approximately 3,100 more lncRNAs than Company N and approximately 2,100 more lncRNAs than Company T (Fig. 4). The UltraMarathonRT libraries seem to exhibit more complexity as they detect similar numbers of protein coding genes with only around half of the total read number.
The additional reads map to introns alongside a large number of unannotated region transcripts that might not have been accessible with retroviral RTs. The retroviral RT reads mostly map to exons, as expected according to previous polyA+ RNASeq research.
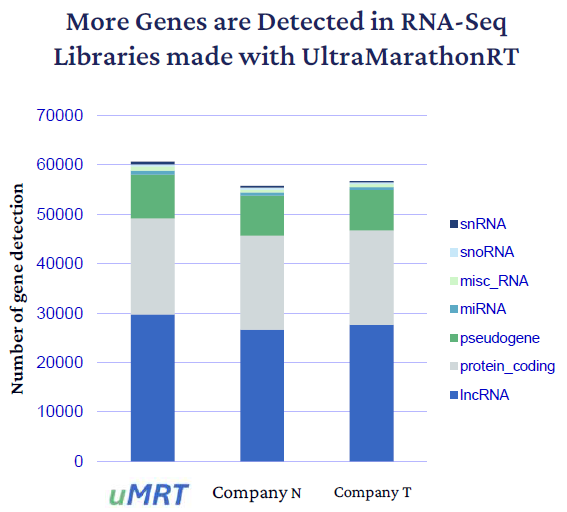
Figure 4. Gene detection across RNA biotypes. UltraMarathonRT detection is higher than retroviral RTs in all RNA classes. The largest gains are observed in noncoding RNAs (blue). Image Credit: RNA Connect
Gene Body Coverage (5’-3’)
UltraMarathonRT enhances gene body (5’ - 3’) coverage by copying each of the transcripts end-to-end in a single pass. Although retroviral RTs delivered a similar extent of gene body coverage for shorter transcripts, uMRT alone displayed proper coverage for transcripts exceeding 2 kb in length.
An observed 5’ bias in the retroviral kits’ may be due to RT extension from internal polyA-rich sites as well as template switching at the 5’ end of mRNA.
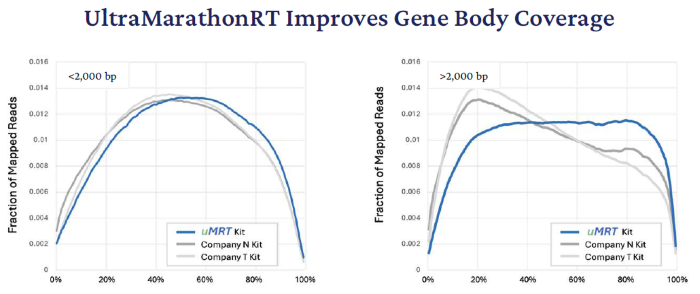
Figure 5. Lines represent the average, normalized transcript coverage uniformity generated using the NCBI curated gene annotation and RSeQC software package. Position 0 % and 100 % on the x-axis represent the 5’ end and 3’ end of the transcripts respectively. Image Credit: RNA Connect
RNA Quantification
As uMRT reliably copies each transcript in an end-to-end fashion in spite of length or structure, it delivers superior quantification, especially for longer transcripts. The retroviral RT kits both underscore the long SIRV transcript spike-ins (4 kb-12 kb) by three to 20 times according to the length.
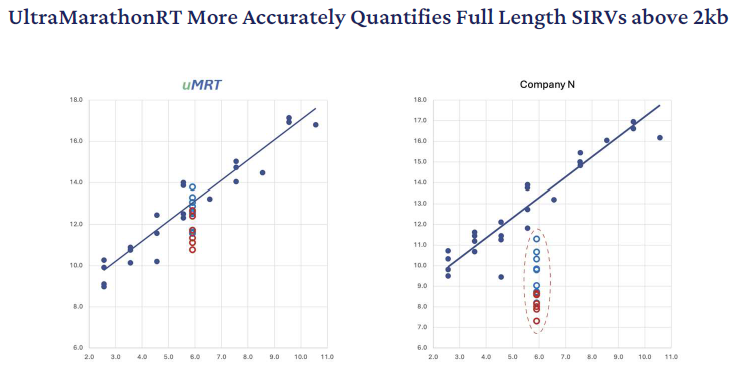
Figure 6. Illumina read pairs from each dataset (18M pairs per sample) were aligned with bowtie2 to the ERCC and SIRV references and transcripts per million reads (TPM) were calculated. Image Credit: RNA Connect
The plots above represent the known molar quantities of the RNA spiked into the original RT reaction (x-axis) versus the observed TPM for each sample. The blue circles represent the 4-8 kb SIRV transcripts. The red circles represent the 8-12 kb SIRV transcripts. The gray dots represent all of the other ERCC and SIRV spike-in transcripts.
To know more about these key variations, RNA Connect visualized read mapping over the longest SIRV spike-in RNA (12 kb) using IGV and saw that the UltraMarathonRT data alone show RNA-Seq reads covering the whole transcript body. This visualization also matches the gene body coverage seen for the endogenous RNAs in the sample.
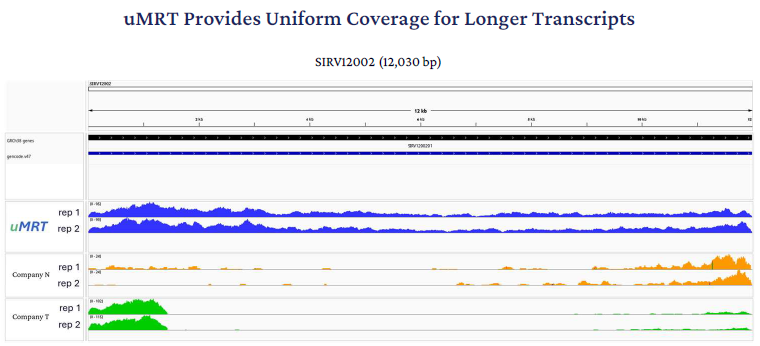
Figure 7. Coverage plots for the longest SIRV Set-4 (Lexogen) Spike-in RNA, SIRV12002, which is 12030 nucleotides in length. The data were generated as spike-in controls in a UHRR Illumina sequencing run; Two replicates are shown for each reverse transcriptase: Blue (uMRT), orange (company N), and green (company T). Image Credit: RNA Connect
Improved Detection of Introns
Intron retention is a widespread biological occurrence that may go undetected owing to the inability of retroviral RTs to make it through the long lengths of multiple eukaryotic introns. As the cDNA synthesis workflow starts with oligo-dT primed cDNA synthesis, intronic reads may start from introns that stay unspliced even following completion of the polyadenylation process..
UltraMarathonRT has been designed to copy through any structure, length, or repeat sequences and so can identify substantially more intron retention events than the retroviral RTs. Matching comparative datasets from the UHRR sample to transcriptomic features demonstrated a significantly improved intron detection when UltraMarathonRT was used for cDNA synthesis.
As this may be an artifact stemming from mispriming, intron detection events were further assessed with IR-FinderS, a special software program built for identifying retained introns in short-read data. To better understand this difference in an independent sample, comparative RNA-Seq datasets were produced with the UltraMarathonRT kit and the same two retroviral kits on two tissue RNA samples: mouse brain and human lung. In both tissues, RNA Connect identified a similar two- to threefold increase in intronic reads.
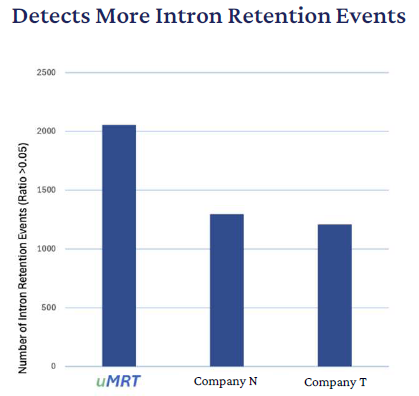
Figure 8. Predicted intron retention events were identified using IRFinder-S’ with default parameters. Pair-ended reads (2x150 bp) from each workflow were used for the analysis. UHRR data analysis with intron retention software: IRFinder-S1. Image Credit: Lorenzi et al. 2021 DOI: 10.1186/s13059-021-02515-8
Summary
The UltraMarathonRT Synthesis and Amplification Kit offers a comprehensive approach for copying polyA+ RNA into cDNA that can be used across multiple applications and analyses. It uses uMRT’s innate helicase activity and ultra-processivity to copy any RNA in an end-to-end fashion within a single pass at ambient temperatures.
This kit’s unique ability was evaluated against top retroviral RT kits on UHRR samples which have been deeply characterized for expression array and RNA-Seq benchmarking.3,4.
Notwithstanding the comprehensive characterization of UHRR, uMRT revealed more genes and retained introns and provided superior gene body coverage and gene quantification.
uMRT revealed new insights into this exhaustively researched sample. The results also indicate the limitations inherent to retroviral reverse transcriptase-based studies and establish the need for superior tools to discover transcriptome data with precision.
References and Further Reading
- Guo, L.-T., et al. (2024). Characterization and implementation of the MarathonRT template-switching reaction to expand the capabilities of RNA-Seq. RNA, (online) 30(11), pp.1495–1512. https://doi.org/10.1261/rna.080032.124.
- Picelli, S., et al. (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature Methods, 10(11), pp.1096–1098. https://doi.org/10.1038/nmeth.2639.
- The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. (2006). Nature Biotechnology, 24(9), pp.1151–1161. https://doi.org/10.1038/nbt1239.
- Li, S., et al. (2014). Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nature Biotechnology, (online) 32(9), pp.915–925. https://doi.org/10.1038/nbt.2972.
About RNA Connect
RNAConnect was founded by innovators in the RNA community for innovators in the RNA community. We think of enzymes as “biotechnology hardware,” and we believe that better enzymes provide more accurate and complete visibility into RNA, which allows you to discover, develop, and innovate faster, easier, and in ways not previously possible.
Sponsored Content Policy: AZoLifeSciences publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of AZoLifeSciences which is to educate and inform site visitors interested in life science news and information.