A landmark Huntington's trial slows disease progression by 75%, signalling how RNA- and CRISPR-based tools redefine treatment for rare genetic disorders.
 Image credit: Vink Fan/Shutterstock.com
Image credit: Vink Fan/Shutterstock.com
Introduction
Significant advancements in gene silencing and gene editing offer new hope for treating rare disorders. Techniques like RNA interference (RNAi) and antisense oligonucleotides (ASOs) can effectively silence defective genes by blocking or altering target messenger RNAs.
Meanwhile, in gene editing, CRISPR-Cas9 allows for accurately correcting disease-causing mutations. Further enhancing CRISPR-Cas9 precision, base editing permits single-letter changes in the nucleotide DNA sequence.
The clinical potential of gene silencing approaches is becoming tangible after encouraging Phase I/II trial data showing a 75% slowing of disease progression in patients treated with AMT-130 gene therapy for Huntington’s disease1. For the millions affected by inherited disorders, these new therapies offer unprecedented hope for truly curative treatments.
Experimental gene therapy slows Huntington's disease in trial | REUTERS
Video credit: Reuters/Youtube.com
Identifying the Problem with Next-Generation Sequencing
Treating a genetic disorder begins with identifying the precise causal mutation, a crucial step that historically subjected patients to a years-long "diagnostic odyssey." Fortunately, this challenge has been overcome by Next-Generation Sequencing (NGS), a high-throughput technology that has dramatically accelerated the process of detecting disease-causing genes.
NGS allows the simultaneous analysis of millions of DNA or RNA fragments, making it possible to rapidly and cost-effectively screen vast stretches of a single genome. Methods such as Whole Exome Sequencing (WES), which focuses on protein-coding regions, and Whole Genome Sequencing (WGS), which examines the entire DNA sequence, quickly pinpoint single-nucleotide variants (SNVs), small insertions/deletions (INDELs), and other complex genetic variations.
The massive volume of data generated by NGS necessitates advanced bioinformatics tools and algorithms for effective analysis, interpretation, and identification of the pathogenic variant. These tools provide functional annotation and help prioritize candidate pathogenic variants2,3. Today the integration of NGS into rare disease pipelines has reduced diagnostic timelines from years to months, enabling faster entry into precision-targeted therapeutic programs.
Turning Off Harmful Genes
Once the defective gene is identified, the next step involves gene silencing, i.e., "turning off" the expression of the faulty gene to prevent the production of a toxic and/or non-functional protein. This is primarily achieved using oligonucleotide-based drugs.
RNA Interference (RNAi)
RNAi is a naturally occurring biological process capable of regulating gene expression. It employs synthetic double-stranded RNA molecules, known as small interfering RNAs (siRNAs). These siRNAs are incorporated into a protein complex that uses one of the siRNA strands to guide itself to the complementary target mRNA. It then cleaves and destroys the target mRNA, effectively silencing the expression of the defective gene.
For example, a recent RNAi-based therapy treats hereditary transthyretin amyloidosis, a fatal disease caused by the accumulation of faulty, misfolded transthyretin (TTR) protein in organs. Patisiran, an RNAi agent, has shown consistent improvements in neuropathy progression and quality of life measures in patients with this devastating disease4.
Similarly, the investigational therapy AMT-130, currently in clinical development for Huntington’s disease, uses an adeno-associated viral (AAV) vector to deliver microRNA that selectively silences huntingtin (HTT) mRNA in brain cells1. Early trial data from UCLH and uniQure have shown a 75% slowing in disease progression over 36 months compared to natural history controls, marking a significant step forward for in vivo gene silencing.
Antisense Oligonucleotides (ASOs)
ASOs are short, synthetic, single-stranded fragments of nucleic acids designed to complement a specific mRNA transcribed from the faulty gene. When the ASO binds to this target mRNA, it physically blocks the cellular machinery from translating the mRNA into a harmful protein or triggers the degradation of the RNA molecule itself.
Multiple ASOs (e.g., eteplirsen, golodirsen, viltolarsen, casimersen) have already been approved by the U.S. Food and Drug Administration (FDA) for treating Duchenne Muscular Dystrophy, each targeting specific exons to restore dystrophin production5,6. Nusinersen (Spinraza), the first FDA-approved ASO for spinal muscular atrophy, further demonstrates how sequence-specific RNA modulation can restore functional protein expression in neurodegenerative disease.
Turning On Gene Expression
Like RNAi, RNA activation (RNAa) is another evolutionarily conserved, sequence-specific mechanism, which differs from the first one in that it upregulates gene expression7,8. RNAa relies on small activating RNAs (saRNAs) that act similarly to siRNAs. This revolutionary approach can be harnessed for treating genetic disorders and advanced gene regulation technologies. The first saRNA drug to reach clinical trials is MTL-CEBPA, which targets the CEBPA gene, a tumor suppressor and regulator of liver function9. While still early in development, RNAa expands the gene regulation toolkit by offering a route to restore underactive or silenced genes, rather than only suppressing overactive ones.
Tackling the Root by Correcting the DNA Itself
While gene silencing offers symptom control by stopping protein production, gene editing aims for a permanent, potentially curative genetic correction by directly modifying the faulty nucleotide DNA sequence present within the patient's genome.
CRISPR-Cas9
The CRISPR-Cas9 system can be defined as a pair of molecular scissors with revolutionary outcomes in precision medicine. It consists of a guide RNA (gRNA) that directs an enzyme (Cas9) to a specific target sequence in the genome. This enzyme then creates a double-strand break in the DNA, and the cell activates natural repair mechanisms to either disable the gene or insert a corrected sequence.
Casgevy (exagamglogene autotemcel) is the first FDA-approved CRISPR/Cas9 therapy. It treats severe inherited blood disorders, including sickle cell disease and transfusion-dependent β-thalassemia, by editing hematopoietic stem cells ex vivo (i.e., outside the human body) to reactivate fetal hemoglobin production. Fortunately, clinical trials have shown high rates of transfusion independence and symptom relief10,11.
This ex vivo approach avoids direct editing inside the body, providing a controlled, high-precision environment before reinfusion of the corrected cell. Additional CRISPR trials are underway for rare disorders such as Leber congenital amaurosis (EDIT-101), a form of inherited blindness, marking the first in vivo CRISPR application in humans12,13.
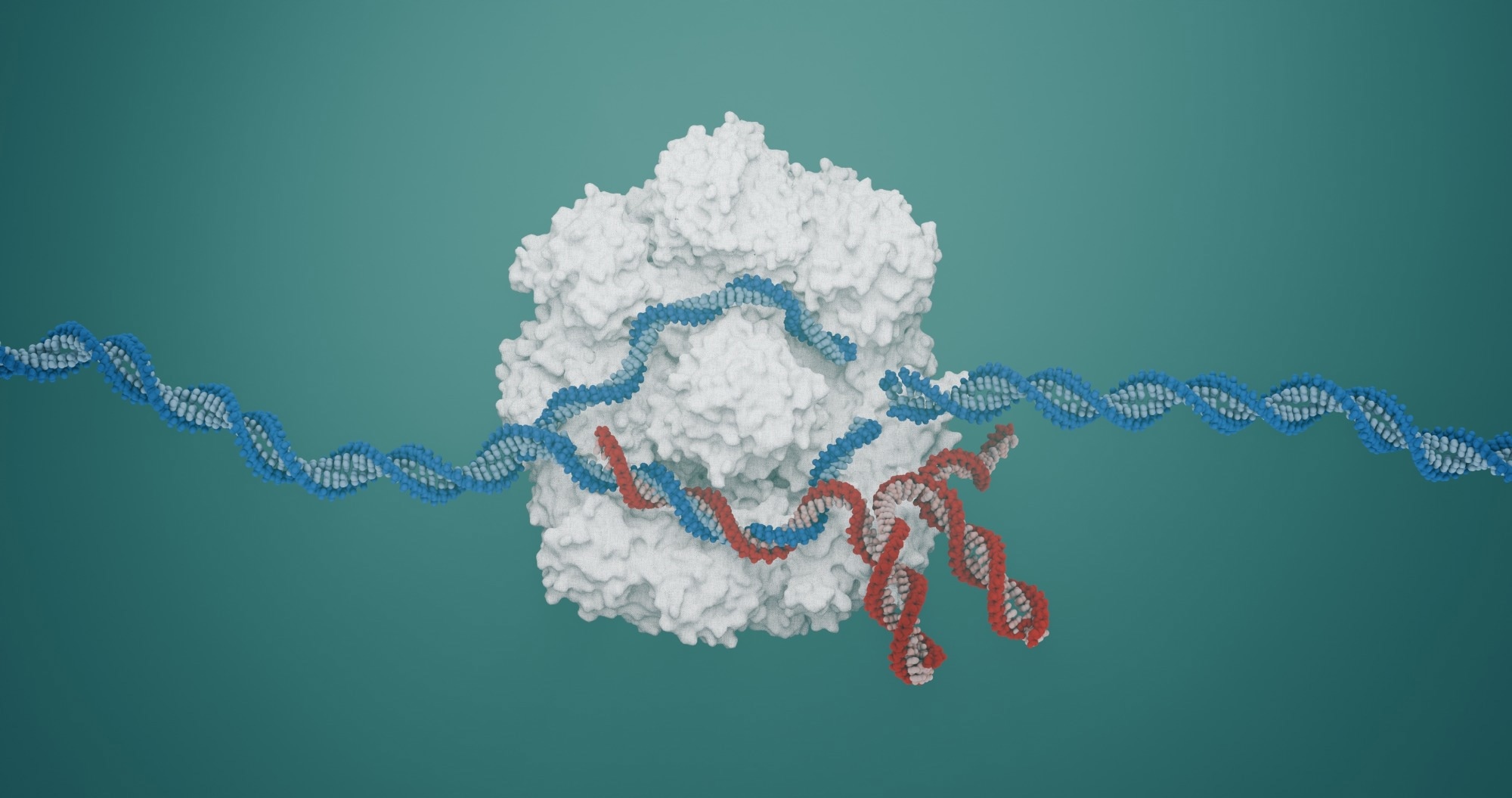 Image credit: Amir Taheri Ghafarokhi/Shutterstock.com
Image credit: Amir Taheri Ghafarokhi/Shutterstock.com
Emerging Editing Technologies
Newer, even more precise gene-editing tools are rapidly advancing, requiring only a lighter touch on the genome:
Base Editing
This enables the change of one single DNA 'letter' (nucleotide) into another without creating a risky double-strand break. This offers huge potential for correcting rare diseases caused by single-point mutations. For example, base editing has shown preclinical correction of the E342K mutation in α1-antitrypsin deficiency models14.
Prime Editing
This can correct all 12 types of single-base changes and also introduce short insertions or deletions. It uses a modified Cas9 enzyme fused to a reverse transcriptase, making it an incredibly versatile tool for gene repair. Though still in early-stage development, prime editing is being investigated for metabolic disorders such as Tay-Sachs and phenylketonuria15.
The Future is Now
The synergy between advanced diagnosis (NGS, bioinformatics tools) and molecular intervention (on/off gene expression and gene editing) is rapidly leading to a new era in treating inherited genetic disorders. From the clinical success of ASOs and RNAi/RNAa to the revolutionary curative potential of CRISPR, these technologies are moving rare disease treatment beyond managing chronic symptoms toward single-administration, root-cause correction.
However, ongoing challenges, including delivery efficiency, immune responses, and equitable access, must still be addressed before these therapies achieve widespread use. As these therapeutic strategies continue to improve, it is realistic to expect that durable, disease-modifying treatments for many monogenic disorders will emerge within the next decade.
References
- Tabrizi, S. J., et al. (2025). Interim results of an AAV-delivered microRNA therapy (AMT-130) for Huntington’s disease: A phase I/II trial update. Nature Medicine, 31(9), 1780–1789. https://doi.org/10.1038/s41591-025-03452-8
- Desvignes, J., Bartoli, M., Delague, V., Krahn, M., Miltgen, M., Béroud, C., & Salgado, D. (2018). VarAFT: a variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Research, 46, W545 - W553. https://doi.org/10.1093/nar/gky471.
- Tuteja, S., Kadri, S., & Yap, K. (2022). A performance evaluation study: Variant annotation tools - the enigma of clinical next generation sequencing (NGS) based genetic testing. Journal of Pathology Informatics, 13. https://doi.org/10.1016/j.jpi.2022.100130.
- Kristen, A., Ajroud-Driss, S., Conceição, I., Gorevic, P., Kyriakides, T., & Obici, L. (2019). Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis.. Neurodegenerative disease management, 9 1, 5-23 . https://doi.org/10.2217/nmt-2018-0033.
- Torres-Masjoan, L., Aguti, S., Zhou, H., & Muntoni, F. (2025). Clinical applications of exon-skipping antisense oligonucleotides in neuromuscular diseases. Molecular Therapy, 33(6), 2689-2704.
- Chen, S., Heendeniya, S., Le, B., Rahimizadeh, K., Rabiee, N., Zahra, Q., & Veedu, R. (2024). Splice-Modulating Antisense Oligonucleotides as Therapeutics for Inherited Metabolic Diseases. Biodrugs, 38, 177 - 203. https://doi.org/10.1007/s40259-024-00644-7.
- Vaschetto, L. M. (2018). Small activating RNAs as promising agents for biotechnological use. Current Pharmaceutical Biotechnology, 19(8), 602-603.
- Vaschetto, L. M. (2018). RNA activation: A diamond in the rough for genome engineers. Journal of Cellular Biochemistry, 119(1), 247-249.
- Setten, R., Lightfoot, H., Habib, N., & Rossi, J. (2018). Development of MTL-CEBPA: Small Activating RNA Drug for Hepatocellular Carcinoma. Current Pharmaceutical Biotechnology, 19, 611 - 621. https://doi.org/10.2174/1389201019666180611093428.
- Laurent, M., Geoffroy, M., Pavani, G., & Guiraud, S. (2024). CRISPR-Based Gene Therapies: From Preclinical to Clinical Treatments. Cells, 13. https://doi.org/10.3390/cells13100800.
- Cetin, B., Erendor, F., Eksi, Y., Şanlioğlu, A., & Sanlioglu, S. (2025). Advancing CRISPR genome editing into gene therapy clinical trials: progress and future prospects. Expert Reviews in Molecular Medicine, 27. https://doi.org/10.1017/erm.2025.10.
- Maeder, M. L., et al. (2019). Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature Medicine, 25, 229–233. https://www.nature.com/articles/s41591-018-0327-9
- Cideciyan, A. V., et al. (2024). Safety and preliminary efficacy of EDIT-101 in Leber congenital amaurosis 10: Results from the BRILLIANCE Phase I/II trial. Investigative Ophthalmology & Visual Science, 65(7), 1445–1454. https://doi.org/10.1167/iovs.65.7.1445
- Wang, Y., et al. (2023). Base editing corrects the E342K mutation in α1-antitrypsin deficiency patient-derived hepatocytes. Molecular Therapy, 31(8), 2620–2631. https://doi.org/10.1016/j.ymthe.2023.03.014
- Anzalone, A. V., et al. (2024). Prime editing in disease models: Expanding the toolbox for therapeutic genome modification. Nature Biotechnology, 42, 1250–1263. https://doi.org/10.1038/s41587-024-01718-1
Last Updated: Oct 16, 2025