Since “brain image capturing” was first developed as early as the 1920s, tremendous advancements have been made. Functional Magnetic Resonance Imaging (fMRI) has been the norm for brain microscopy since it came to fruition in the 1970s, and it is here that humanity has hit a brick wall of sorts. Only in the past 10 years have our microscopy practices been evolving in terms of sensitivity, and tunability.

Image Credit: E-ART/Shutterstock.com
Recent innovations in microscopy have shed new light on the connectome, and neural mapping. These recent innovations take the form of single-photon microscopy, two-photon microscopy, and multi-scale light-Sheet Fluorescence Microscopy.
Why it is difficult to take images of the brain
Neurons themselves correspond through ion channels; vacillating charge gradients which change the chemical energy within our system into kinetic energy. This is done at the micro-scale (1*10-9m) and is impossible to capture through our current approaches. Many have tried to circumvent this issue of size and scale, but with 100 billion neurons in the brain, each connected to thousands of other neurons, all creating’s thousands of trillions of synapses, the math simply cannot be done.
“Neuronal Noise” was first observed in 1953 by Stephen W. Kuffler. This broadens the scope of view by using an alternate analytical signal (noise rather than sight), though the same problem of magnitude still presents itself. The theoretical map out of all these neurons and their adjoining circuits has been christened as “the connectome”, a nebulous cloud that we one day hope to understand completely.
Many can contest that fluorescence microscopy is what founded this new frontier in functional imaging, optimized only recently in the past decade or so. Single-photon and two-photon fluorescence microscopies are what encompass this field, each with its own suite of gains and applications.
Single Photon-Wide Field Fluorescence Microscopy
Conventional microscopy, or single-photon microscopy, emits from a range of ultraviolet light to blue light within the visible spectrum. In the scope of brain imaging, these wavelengths will excite fluorescent molecules that emit from an inert dye. Widefield fluorescence microscopy can be used in this way and can supply us with an image of the brain in its entirety.
The brightness of this signal is dependent on the light-condensing power of the object that is being illuminated (∝NA2), and the light-gathering power of that object (∝NA2) (NA=numerical aperture). and the magnification of the whole image (∝1/Mag2). This is the most common derivative of microscopy, where blue or ultraviolet light illuminates, yielding a green emission from GFP (green fluorescent protein) which we can then measure.
This methodology can fall short however when assaying neural tissue. Implementing light that is lower down the electromagnetic spectrum (increasing in frequency) will result in a blurred image if used on milky tissue. Here is where two-photon microscopy holds an advantage.
Two-Photon Microscopy in Neural Imaging
Spacing out at less than a trillionth of a second, two-photon microscopy excites these fluorophores using two photons in a single event. Given that the intensity of the emitted infrared light is so much stronger than that of UV, it must pulse at infinitesimally brief intervals. Two photons are used because energy is inversely proportional to wavelength, as is dictated by wave, particle duality.
What this means is that two photons must be used to achieve the same energy as a single photon microscope. This is a highly specific procedure, meaning that to achieve excitation the photons much reach their target loci within 1*10-18 of each other.
Exciting the fluorophores via red/infrared light can achieve a resolution that scales with single cells, allowing for optimal in vivo imaging of the brain. This is because red light/infrared light has a smaller wavelength, equating to higher energy which can penetrate deeper within tissue and results in less light scatter. It can penetrate up to half a millimeter below the surface resulting in nonlinear absorption, meaning it is proportional to the amount of light per square inch (optical intensity).
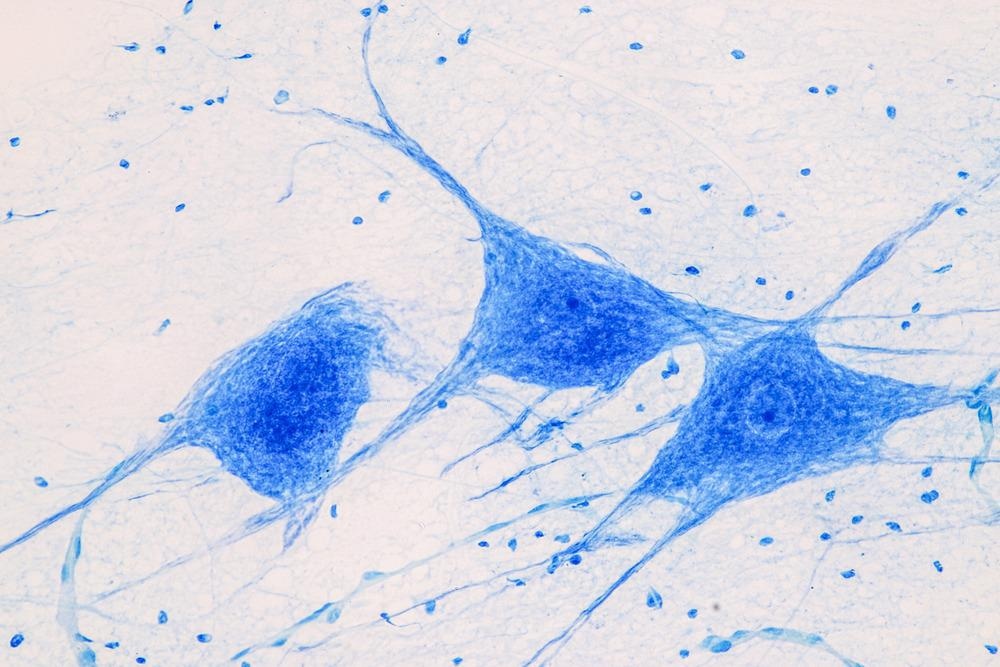
Image Credit: Rattiya Thongdumhyu/Shutterstock.com
Multi-Scale Light-Sheet Fluorescence Microscopy
Light-sheet fluorescence microscopy (LSFM) encompasses an even more optimized version, tuned specifically for neural imaging. This apparatus will dissociate the two optical paths of detection and illimitation using a thin plane of light. This light is perpendicular to the optical path of detection, equating its image luminescence to (NA2/Mag2), as opposed to wide-field microscopy (NA4/Mag2). This apparatus is fit with automated zooming, precise intervals of sectioning, and extreme tissue expansion. The smaller ratio of numerical aperture vs. magnification is to blame for this.
Using mouse models, researcher labs have been able to map out electrode tracks, axonal projections, and potential energy gradients. Research labs, like the team of Zengcai V. Guo et al., track these reactions and species at the sub-micron level and are elucidate the morphology of the prefrontal cortex neurons (the region of the brain responsible for cognitive control functions, dopamine levels, etc.)
We still have a long way to go before viewing the entire connectome, which may be accomplished within our lifetime with the implementations of deep tech and AI. As for our current paradigm, these varying devices carry pros and cons depending on the scope of view, and whether the assays are in vivo or in vitro.
Find out more about fluorescence microscopy by clicking here.
Sources:
- Zhang Z, Yao X, Yin X, Ding Z, Huang T, Huo Y, Ji R, Peng H, Guo ZV. (2021) Multi-Scale Light-Sheet Fluorescence Microscopy for Fast Whole Brain Imaging. Front Neuroanat. 24;15:732464.
- Alivisatos, A. P., Schnitzer, M., Sejnowski, T. J., … Zhuang, X. (2013). Nanotools for neuroscience and brain activity mapping. ACS nano, 7(3), 1850–1866.
- Sanderson, M. J., Smith, I., Parker, I., & Bootman, M. D. (2014). Fluorescence microscopy. Cold Spring Harbor protocols, 2014(10), pdb.top071795.
- Hudson, D. E., Hudson, D. O., Wininger, J. M., & Richardson, B. D. (2013). Penetration of laser light at 808 and 980 nm in bovine tissue samples. Photomedicine and laser surgery, 31(4), 163–168.
- O'Shea, D. J., Trautmann, E., Chandrasekaran, C., Stavisky, S., Kao, J. C., Sahani, M., Ryu, S., Deisseroth, K., & Shenoy, K. V. (2017). The need for calcium imaging in nonhuman primates: New motor neuroscience and brain-machine interfaces.
- Experimental neurology, 287(Pt 4), 437–451. https://doi.org/10.1016/j.expneurol.2016.08.003
Further Reading
Last Updated: Mar 4, 2022
 Scientists Track Tiny Human Proteins Using Novel Laser Phase Plate
Scientists Track Tiny Human Proteins Using Novel Laser Phase Plate