A major proteomics review shows how tracking shifts in protein structure, not just protein levels, can uncover hidden biology, sharpen drug-target discovery, and open new paths for structural biomarker research.
Review: 3D Proteomics: Structural, Functional, Chemical and Biomarker Discovery Proteomics With LiP-MS. Image Credit: Vink Fan / Shutterstock
In a recent review published in the journal Molecular & Cellular Proteomics, researchers at the Institute of Molecular Systems Biology, ETH Zurich, Switzerland, explained how limited proteolysis coupled to mass spectrometry (LiP-MS) enables proteome-wide detection of protein structural changes and their biological significance.
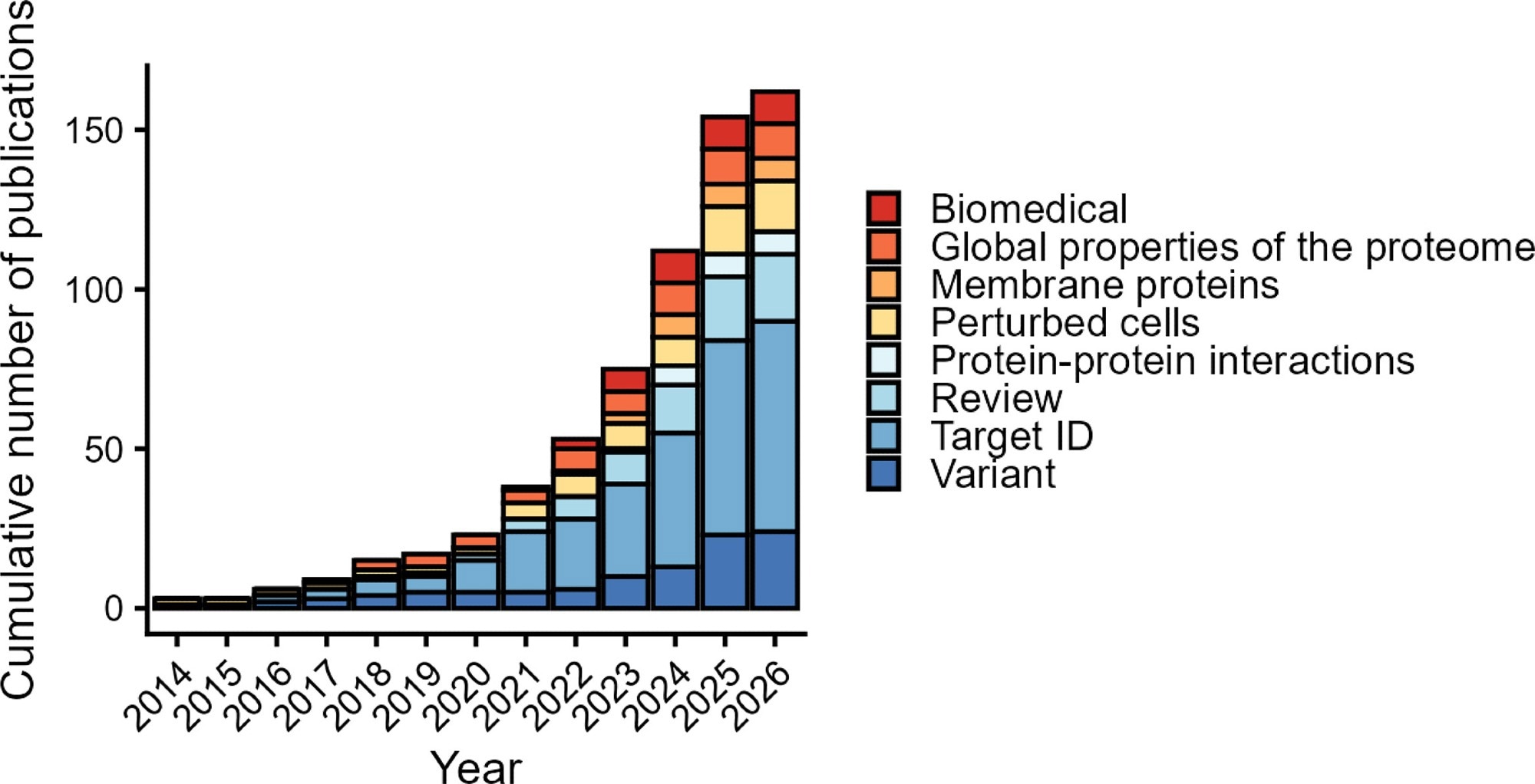
LiP-MS is being widely applied. The plot shows the cumulative number of publications using LiP-MS or a variant in every year since the method was described (6) until submission of the revised version of this review. Broad categories of applications or methodological variants are indicated with color. We note that studies where LiP-MS was applied to single purified proteins were not included in this graph.
Protein Structure Changes Beyond Abundance
What if two cells had the same proteins but behaved completely differently? It is often not about protein amount but rather protein structure. Traditional proteomics mainly measures protein abundance, missing dynamic structural changes that control function.
Proteins constantly change structure due to stress, drugs, or disease, influencing how cells behave, and identifying these changes is essential for understanding health and disease mechanisms.
Structural proteomics methods aim to address this gap, offering deeper insight into cellular regulation. However, current approaches still face limitations in resolution and coverage, so more comprehensive, system-wide techniques are needed.
Proteins are not static entities; they behave more like flexible machines that constantly change shape in response to their environment. These changes determine how proteins interact, perform enzymatic reactions, and regulate essential processes. For example, a drug can alter a protein's shape, potentially affecting its overall function. Similarly, diseases such as neurodegenerative disorders often involve misfolded proteins.
Although protein quantity in a sample can be measured using conventional proteomics techniques, that alone is often insufficient to explain functional differences. Structural information provides deeper insights, making it important in medicine, drug design, and biotech.
LiP-MS Principles and Method Variants
LiP-MS is a technique designed to detect these structural changes at a large scale. It works on a simple yet powerful principle: when proteins are exposed to a protease enzyme for a short time, the enzyme cleaves accessible, flexible regions of the protein. When a drug or stress alters a protein's shape, the cleavage pattern also changes, and mass spectrometry detects these differences.
Researchers extract proteins, then treat them with Proteinase K for a short time, and then analyze the broken-down pieces. By comparing treated and untreated samples, researchers can identify structural differences among proteins, providing a broad view of protein structural dynamics rather than focusing on just one.
Over time, several variations of LiP-MS have been developed to improve sensitivity and applicability. Thermolysin or trypsin can be used based on the requirement. Some methods introduce ligands in varying concentrations to identify drug targets more precisely, such as the LiP-Quant approach. Others, such as in-cell LiP-MS, allow structural analysis directly within living cells, thereby preserving more native cellular conditions. The review argues that LiP-MS enables a new form of “3D proteomics,” adding protein structural information to traditional proteomics approaches that mainly track protein abundance.
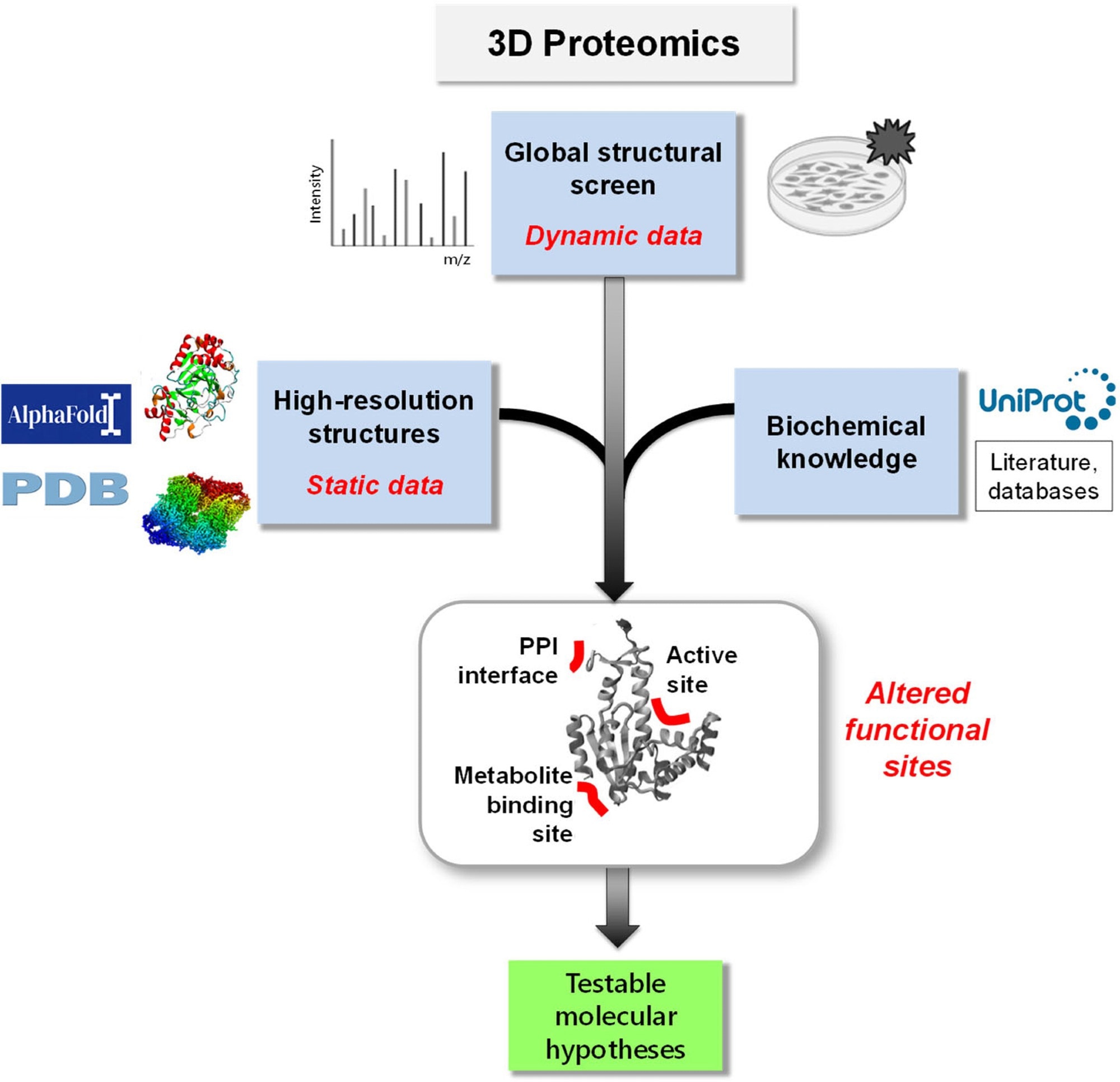
3D Proteomics. Integrating proteome-wide structural dynamics data (such as LiP-MS data) with high-resolution 3D protein structures and prior biochemical knowledge constitutes a new type of functional omics screen. 3D proteomics generates a wealth of molecular hypotheses about structural and functional changes in individual proteins with peptide-level resolution and much enriches the systems-wide functional information that can be obtained in proteomics screens.
These advancements make the method adaptable for diverse biological questions. For instance, enrichment techniques can increase detection sensitivity, while computational tools help distinguish structural changes from simple abundance differences. The review also emphasizes that different variants involve tradeoffs in sensitivity, coverage, reproducibility, and analytical complexity.
LiP-MS Applications in Disease and Drug Discovery
LiP-MS has broad potential and can be used in numerous ways, including studying cell responses to environmental stresses and identifying structural changes that take place before any measurable changes in protein abundance in the cell. These data are important for understanding cells' responses to rapid environmental changes.
LiP-MS has revealed disease-associated structural changes in conditions such as Parkinson's and Alzheimer's diseases. These changes can serve as candidate “structural biomarkers” and help researchers explore new ways to detect and understand disease-related dysfunction. Rather than using traditional biomarkers, which measure the concentration of proteins in cells, structural biomarkers provide a deeper level of information regarding the disruption of protein function.
This method is also very useful in drug discovery. By observing how proteins change structure upon binding to small molecules, researchers can identify candidate drug targets and understand how they work. For example, LiP-MS has been used to identify interactions between proteins and metabolites, drugs, and even nanoparticles, which may support the development of new, more effective therapies. However, such target discoveries often still require orthogonal validation.
Global Proteome Insights from LiP-MS
Beyond individual proteins, LiP-MS provides insights into the global properties of the proteome. It identifies proteins that are more stable, which are prone to aggregation, and how proteins respond to extreme conditions like heat or pressure. These findings are important for understanding cellular resilience and adaptation.
LiP-MS has also shown that many proteins undergo structural changes under stress without corresponding changes in abundance. This supports the use of structural data alongside traditional methods because it can reveal functional alterations that abundance measurements alone may miss, including changes linked to aging and disease processes.
Future Directions for Structural Proteomics
Despite its advantages, LiP-MS has limitations, as it requires sufficient protein coverage, meaning some regions or low-abundance proteins may be missed. The large number of peptides generated also complicates data analysis. Nevertheless, improved computational tools and experimental design are addressing these difficulties.
In the future, integrating LiP-MS with other technologies, such as machine learning and multi-omics approaches, could further enhance its capabilities. This might allow researchers to predict protein behavior more precisely, making it easier to explain complex biological systems.
LiP-MS has enhanced proteomics by shifting the focus from protein quantity to protein structure. It shows how proteins behave in complex biological samples, uncovering functional changes that traditional methods often miss. This method has significant implications for drug development, identification of disease biomarkers, and understanding mechanisms of regulation, and therefore has important potential applications in healthcare and biotechnology.
Although challenges remain, future advances in structural proteomics, along with the increasing use of high-throughput techniques, may enable it to play a crucial role in the modern biological sciences community.
Proteome-level understanding of protein structure will ultimately support disease research, biomarker discovery, and the development of molecular hypotheses about how biological systems operate.
 Decoding Hidden Metabolic Tissue Crosstalk via Advanced Proteomic Tracking
Decoding Hidden Metabolic Tissue Crosstalk via Advanced Proteomic Tracking