A head-to-head comparison of four proteomics workflows shows how next-generation DIA platforms can detect far more of the proteome, sharpen biological insights, and cut sample needs in a widely used mouse model of lung injury.

Study: What Does Next-Generation Mass Spectrometry Offer for Proteomics? A Comprehensive Platform Comparison. Image Credit: Sodel Vladyslav / Shutterstock
A recent study published in the Journal of Proteome Research shows that next-generation mass spectrometry platforms are reshaping proteomic analysis.
Comparing advanced systems such as the Orbitrap Astral and timsTOF Ultra with the Orbitrap Exploris 480 in a neonatal mouse model of bronchopulmonary dysplasia (BPD), researchers found that data-independent acquisition (DIA) workflows, especially on newer instruments, significantly improve protein and peptide detection.
These gains enable more complex biological insights, stronger statistical power, and fewer required samples, enhancing systems-level understanding without introducing bias and accelerating more cost-effective disease research, a meaningful step toward more efficient biomedical discovery.
Proteomics Background and DIA Platform Gap
Bottom-up proteomics remains a cornerstone for high-throughput protein characterization, traditionally relying on data-dependent acquisition (DDA). More recently, DIA has emerged as a powerful alternative.
Advances in instrumentation and bioinformatics have improved DIA’s depth, but challenges in data complexity and cross-platform consistency limit broader adoption.
Recently introduced next-generation systems, such as the Orbitrap Astral and timsTOF Ultra, promise considerable gains in sensitivity, speed, and proteome coverage. However, most benchmarking efforts have evaluated these platforms in isolation or against legacy instruments. This leaves a critical gap in direct, multivendor comparisons under optimized but comparable experimental conditions.
Mass Spectrometry Benchmarking Study Design
In the present study, researchers at Case Western Reserve University, USA, designed a comparative benchmarking study to evaluate the global proteomic performance of four mass spectrometry workflows.
They analyzed neonatal mouse lung tissues from a BPD model (n = 12), randomizing animals into normoxia (21% O2) and hyperoxia (85% O2) groups for 14 days. They then euthanized the animals and aliquoted samples to ensure the same biological material was analyzed across platforms, even though LC columns, gradients, and sample loads were not identical.
The team compared the Orbitrap Exploris 480 in both DDA and DIA modes with two next-generation systems, the Orbitrap Astral (HR-DIA) and timsTOF Ultra (DIA-PASEF). They performed label-free quantitative proteomics, using standardized sample preparation but platform-optimized liquid chromatography-mass spectrometry (LC-MS) analysis.
To benchmark performance, the investigators assessed proteome depth using precursor, peptide, and protein identifications at a 1.0% false discovery rate (FDR). They then examined the influence of increased proteome coverage on biological interpretation through subcellular localization, pathway enrichment, and systems-level analyses.
The authors compared protein expression between normoxia and hyperoxia using linear modeling with empirical Bayes moderation to evaluate biological relevance. They used principal component analysis (PCA) and correlation testing to assess data structure and cross-platform concordance.
Gene Ontology (GO) and pathway enrichment analyses, alongside targeted evaluation of disease-relevant proteins, provided functional insights. Lastly, the researchers estimated statistical power and sample size requirements to determine whether enhanced proteome coverage could reduce the number of biological replicates needed.
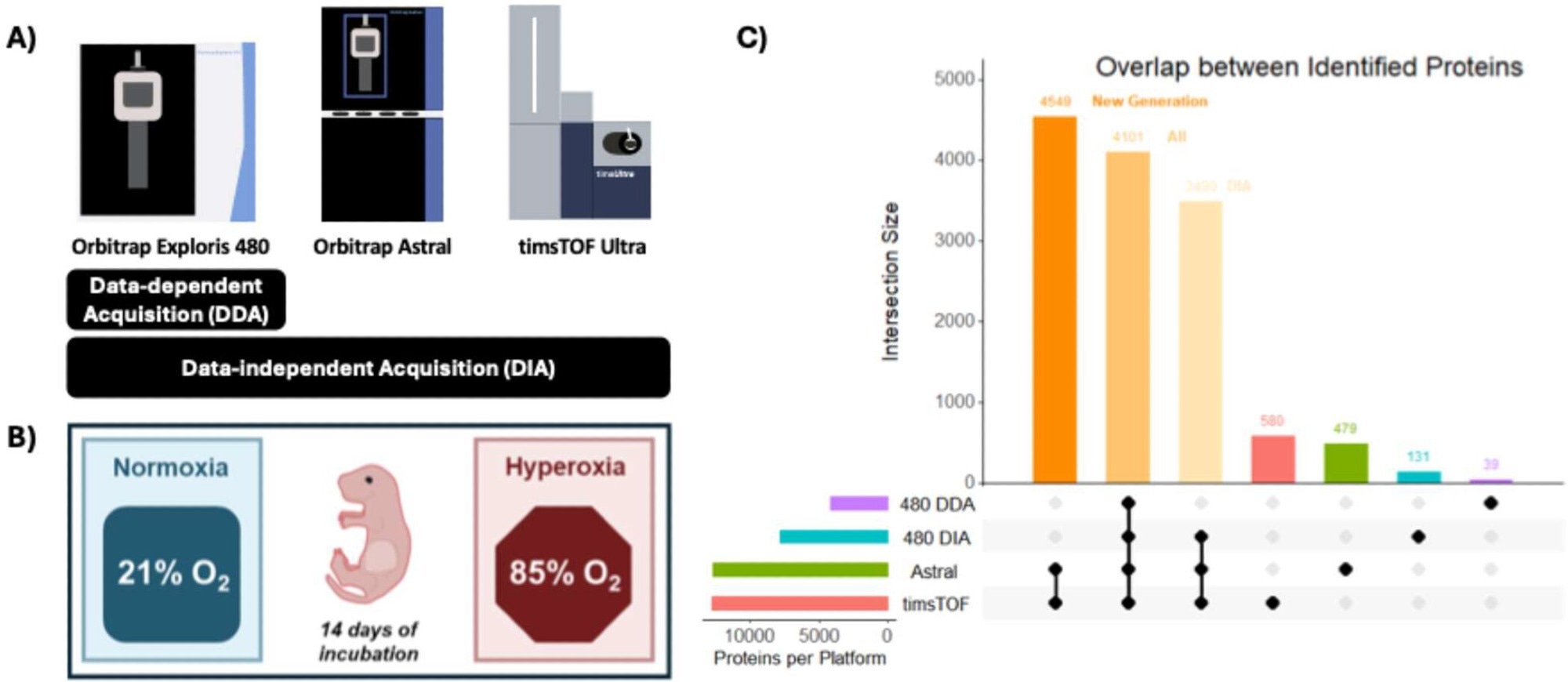
Study scheme and BPD model. (A) Schematic overview of MS instrumentation and data acquisition mode. (B) Mouse pups were incubated for 14 days in either normoxia or hyperoxia conditions. (C) Upset plot highlights the overlap among proteomes from different platforms (Orbitrap Exploris 480 DDA in purple, Orbitrap Exploris 480 DIA in blue, Orbitrap Astral in green, and timsTOF Ultra in coral). Mouse pup illustration (B) was sourced from SciDraw and adapted from Heath Robinson (licensed under a CC-BY 4.0 license).
Next-Generation DIA Performance Results
The results reveal a striking expansion in proteome coverage with DIA workflows and next-generation platforms, without compromising biological consistency. Across all workflows, more than 4,000 proteins were commonly identified, with DIA recapitulating over 98–99% of proteins detected by traditional DDA. However, the depth of analysis diverged sharply: the timsTOF Ultra and Orbitrap Astral quantified more than 225,000 peptides and more than 13,000 proteins, representing up to an eightfold increase in peptide depth and over threefold higher protein coverage relative to the DDA workflow on the Exploris 480.
The increased sensitivity translated into richer biological insights. Subcellular compartment coverage rose from 30% with the DDA approach to 66% with next-generation systems, while pathway annotation improved from 58% to 90%.
Notably, DIA datasets revealed a markedly higher number of differentially expressed proteins associated with phenotype-related changes, with up to a fourfold increase compared to DDA. Many were linked to key cellular components, including mitochondria, ribosomes, and extracellular regions central to disease biology.
Despite this expansion, functional analyses showed no bias in functional annotation of newly detected proteins, suggesting that DIA enhances completeness rather than altering biological interpretation.
The improved data quality also strengthened statistical performance. PCA showed clearer separation between experimental groups, with variance explained increasing from 28% (DDA) to 46% on next-generation platforms. At the same time, sample size requirements dropped dramatically, from 15 replicates per group with DDA to just four using timsTOF Ultra, an approximately 66% reduction. High concordance across platforms (r = 0.87) further underscored the robustness of these findings, positioning next-generation DIA workflows as both deeper and more efficient for proteomic studies.
Future Proteomics Applications and Implications
The findings highlight a clear shift in proteomics toward data-independent acquisition on next-generation platforms, where improved sensitivity and ultrafast scan rates enable deeper, more comprehensive proteome profiling. This expanded coverage not only enhances biological interpretation but also strengthens statistical power, considerably reducing sample size requirements in this experimental model.
While some variability persists in differential expression analyses and technical differences between platforms complicate direct comparisons, the overall concordance underscores their reliability. Looking ahead, continued advances in instrument design, ion optics, and bioinformatics are likely to further refine these capabilities.
As these technologies mature, they are poised to support emerging applications such as spatial proteomics and integrative multi-omics, driving more precise and systems-level insights into complex biological processes.
 Study Shows How AI and Single Cell Science are Changing Modern Medicine
Study Shows How AI and Single Cell Science are Changing Modern Medicine